В статье расскажем всё о синдроме Ангельмана: определение, историю, причины, симптомы, приведём пример из практики с фото и видео. По каким признакам узнать и как лечить болезнь Ангельмана.

Синдром Ангельмана – это хромосомная патология, локализованная в 15 хромосоме, проявляющаяся выраженной задержкой психо — речевого и моторного развития, расстройством координации, нарушением поведения из группы расстройства аутистического спектра, особенностями эмоциональной сферы («счастливое» выражение лица в сочетание со вспышками смеха, улыбчивость, доверчивость), стереотипиями (взмахи или похлопывания руками), эпилептическими приступами.
Отличительные признаки синдрома Ангельмана
Марионеточная походка, приступы смеха, радостное лицо.
Распространенность синдрома Ангельмана 1 на 10-30 тысяч новорожденных. Но большое количество случаев остаются не диагностированными, а наблюдаются у невролога с задержкой речевого развития, нарушениями поведения, эпилепсией. Мальчики и девочки болеют с одинаковой частотой. При изучении частоты встречаемости среди пациентов с умственной отсталостью выявлено 4,8% больных синдромом Ангельмана.
История
Заболевание названо по имени детского врача из Британии Гарри Ангельмана.

В 1965 году Эйнджелмен (вариант написания) описал трёх мальчиков из разных семей, назвал «Синдром счастливой марионетки» или «happy puppet syndrome», по их ангельскому счастливому внешнему виду и приступам беспричинного смеха, резким движениям в руках, дёрганной марионеточной походкой, тяжелой умственной отсталостью. Аналогия была проведена Эйнджелменом, глядя в музее Castelvecchio Вероны на картину «Мальчик – марионетка» с изображением смеющегося мальчика.

Чарльз Уильямс и Джейми Фриас в 1982 году описали ход болезни и предложили по этическим соображениям термин «синдром счастливой марионетки» заменить по имени описавшего педиатра в Синдром Ангельмана (СА).
Факты сегодняшней жизни
Среди популярных людей в настоящее время имеют в своей семье детей с Синдромом Ангельмана: Актер Колин Фаррелл, автор Иан Ранкин, профессиональный бейсболист Дэйв Хендерсон, профессиональный хоккеист Питер Мак Дафф.

Ирландский киноактер Колин Фаррелл в 2007 году поделился, что у их сына верифицирован диагноз СА. Как преданный отец Colin James Farrell любит и заботится о сыне, а также оказывает моральную поддержку для подобных семей с детьми — инвалидами. Он участвует в FAST – фонде терапии синдрома Ангельмана. 6 декабря 2013 года в Чикаго был благотворительный Гала вечер Фонда СА, на котором принял участие К. Фаррелл.
Этиологии патогенез
Причина синдрома Ангельмана: в геноме недостаёт части из 3 — 4 миллионов пар оснований ДНК короткого плеча q11—q13 материнской копии пятнадцатой хромосомы.

Повреждение 15 хромосомы даёт два варианта патологии: при дефекте материнской хромосомы ребенок рождается с синдромом Ангельмана, при дефекте хромосомы отца – с синдромом Прадера — Вилли.
Выявлено четыре генетических варианта появления синдрома Ангельмана:
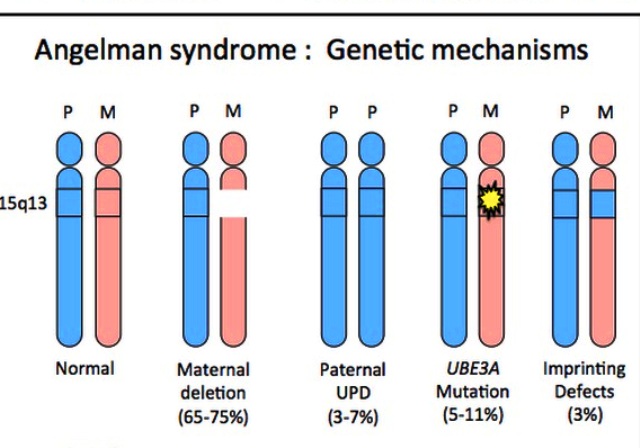
- Мутация вновь возникшая – делеция в локусе 15 q11—q13 составляет большинство до 80 % всех случаев болезни.
- Диссомния по отцовской линии при потере передачи материнского локуса составляет 5% от всех случаев.
- Дефект центра импритинга до 5 %.
- Спонтанная мутация материнской копии вызывает отсутствие экспрессии родительской копии гена UBE3A в мозге. UBE3A кодирует деятельность фермента E6-AP убиквитин лигазы, которая выбирает один из четырех субстратов Е6-AP. Дефицит фермента E6-AP убиквитин лигазы, входящего в сложный процесс распада белков в гиппокампе – молекулярный механизм синдрома.
- У 7-9 % генетический синдром не идентифицируется.
Риск повторного рождения детей больных этим синдромом при вновь возникшей мутации в одной семье низкий — около 1%. Но в крайне редких случаях риск до 50%, если мутация в центре импритинга.
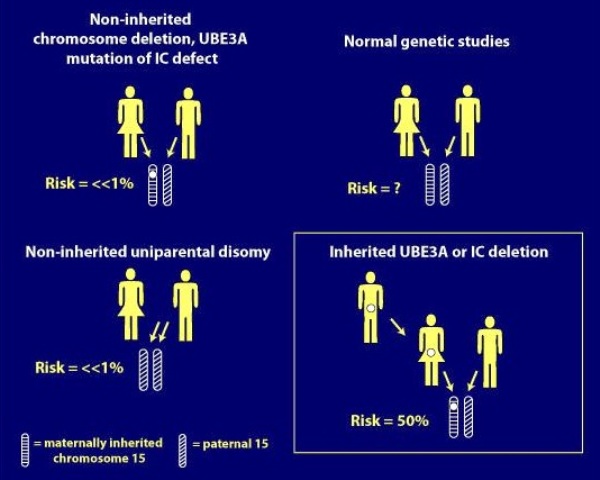
Тип генетической передачи определяет тяжесть клинических проявлений: при делеции делеция в локусе 15 q11—q13 (1 вариант) более грубые умственные и двигательные нарушения, резистентные к терапии эпилептические приступы.
При этом виде мутации происходит снижение ГАМК – рецепторов типа А.
Критерии диагноза синдрома Ангельмана
Симптомы, полезные в качестве поддерживающих критериев, но отклонение от них не исключает диагноза СА:
Нормальное течение перинатального периода; нормальная окружность головы при рождении; отсутствие выраженных пороков развития.
- Очевидная задержка развития, начиная с возраста 6- 12 месяцев жизни.
- Отсутствие прогрессирующей утраты приобретённых навыков.
- Нормальные результаты метаболических, гематологических и биохимических лабораторных тестов.
- Отсутствие структурных изменений в головном мозге по данным МРТ или КТ. Часто описывают умеренную кортикальную атрофию, проявление дисмиелинизации.
Обязательные симптомы — встречаются в 100 % случаев при СА:
- Выраженная задержка психического развития.
- Речевые нарушения: отсутствие речи или скудный словарный запас (не более 6 слов).
- Возможно невербальное общение.
- Атаксия (это расстройство координации)
- Тремор конечностей
- Специфические особенности поведения: гиперактивность, стереотипии в виде размахивания руками; частый беспричинный смех.
Часто встречающиеся симптомы (у 80 %)
- Постнатальная микроцефалия (окружность головы при рождении менее 32 см, к 1 году около 42 см)
- Эпилепсия
- По ЭЭГ феномены: в виде высокоамплитудных разрядов медленных комплексных острая — медленная волна частотой 2-3 Гц, часто провоцируется закрывание глаз.
Дополнительные критерии — у 20-80 %
- Плоский затылок с канавкой.
- Высунутый язык. Отсюда и проблемы при сосании, жевании.
- Слюнотечение.
- Широкий рот с широкими редкими зубами.
- Прогнатия (это выступающая вперед нижняя челюсть).
- Косоглазие.
- Гипопигментация кожи, светлые волосы и глаза (только в случае делеции).
- Усиления сухожильных рефлексов с ног.
- Приподнятые плечи и полусогнутые в локтевых суставах руки при ходьбе.
- Плохая переносимость душных помещений.
- Нарушения сна.
- Повышенное внимание и притяжение к воде.
- В ответ на поднесённый к уху звучащий камертон больные широко улыбаются, смеются и наклоняются к камертону. Это может служить дополнительным диагностическим признаком при осмотре больных старше 1 года.
Клинические проявления синдрома Ангельмана
Внешний вид людей с синдромом Ангельмана.
Особенный черепно-лицевой и скелетный дисморфизм:
- Микроцефалия
- Гипоплазия средней части лица
- Глубоко посаженные глаза
- Выступающая вперед нижняя челюсть
- Протрузия языка (язык высунут и широкий)
- Заострённый подбородок
- Широкие межзубные промежутки
- Часто бледная кожа, светлые волосы, голубые глаза
- Укорочение сухожилий
- Сколиоз
Часты соматические и вегетативные расстройства: склонность к запорам, пищеводный рефлюкс, диффузная гипотония мышц, плохая переносимость жары.
При проведении неврологического осмотра выявляются:
Косоглазие, диффузно сниженный мышечный тонус, повышение сухожильных рефлексов с конечностей, атаксия. Задержка моторного и психо — речевого развития умеренной и тяжелой степени тяжести.
Походка
Специфическая атаксическая походка: как марионетка – кукла на канатиках; «дерганная».
Резкие движения руками помогают удерживать тело в пространстве, балансировать.
Гиперкинезы:
Стереотипии — характерны необычные для здоровых повторяющиеся движения в виде взмахов рук, хлопанье в ладоши, кручения кистями.
Неэпилептический миоклонус — подергивания (вздрагивания) в конечностях.
Легкий тремор в покое.
Как распознать синдром Ангельмана
Частые симптомы синдрома Ангельмана:
Психомоторное развитие.
- Тяжелая умственная отсталость с выраженной задержкой двигательного развития — 100%
- Приступы немотивированного смеха
- Отсутствие речи или минимальный словарный запас (менее 5-6 слов) — 100 %
Голова
- Микроцефалия
- Светлые волосы — 65 %
- Глубоко посаженные глаза
- Аномалия глаз, включая гипопигментацию сосудистой оболочки и радужки, у 88 % голубые глаза.
- Косоглазие 42 %
- Верхние микрогнатия
- Большой рот с высунутым языком и широкими межзубными промежутками
- Нижняя прогнатия
Центральная нервная система
- Атаксия
- Специфические движения рук и ног при ходьбе, напоминающие движения марионетки -100%
- Характерное положение рук: поднятые и согнутые в локтевых и лучезапястных суставах.
- Мышечная гипотония
- Гиперрефлексия
- Леворукость
- Эпилептические припадки:
- Генерализованные тонико-клонические судороги, инфантильные спазмы, приступы падений, абсансы, миоклонические приступы, статус абсансов, конвульсивный статус, сложные парциальные приступы, полиморфные приступы (2 и более видов приступов).
- Возраст дебюта от 3 месяцев до 20 лет, чаще начинающихся в 18- 24 месяцев (86%).
- Начало часто с фебрильных приступов (50%). У детей старшего возраста приступы учащаются при повышении температуры тела выше 37,5 С до серийных приступов или эпилептического статуса.
ЭЭГ
- высокоамплитудные пики и медленные волны с частотой 2- 3 Гц преимущественно в лобных отделах,
- высокоамплитудные медленные волны,
- пики в затылочных отделах, усиливающиеся при закрытых глазах,
- генерализованная высокоамплитудная медленная активность на протяжении почти всей записи 92%.
На МРТ и КТ картина неспецифическая:
- атрофия больших полушарий (33 %), расширение сильвиевых борозд, нейрональные гетеротопии, атрофия мозжечка, задержка миелинизации.
Кожа
- Гипопигментация (39 %)
Редкие симптомы
- Сколиоз, близорукость, дальнозоркость, нистагм.
Течение и прогноз при Синдроме Ангельмана
- Умственная отсталость не прогрессирует, но остаётся очень тяжелой.
- Эпилептические припадки особенно частые и тяжелые в возрасте около 4 лет, а к 10-14 годам могут прекратиться.
- Смех вызван не положительными эмоциями, а поражением на уровне ствола мозга.
- У больных снижена потребность во сне, особенно в возрасте 2-6 лет.
- Несмотря на отсутствие речи, они могут общаться жестами.
- Развивается способность выполнять простые словесные инструкции.
- Некоторые могут пользоваться горшком к 4-5 годам.
- Каждый с СА не способен жить без постоянного ухода.
Лечение Синдрома Ангельмана
Медикаментозное лечение синдрома Ангельмана состоит:
- Противоэпилептической терапии;
- Коррекция поведенческих нарушений;
- Улучшения сна;
- Помощь в овладении навыками (двигательными, речевыми, по уходу);
- Адаптивная физическая культура.
Как общество относится к инвалидам, для чего необходимо адаптивное физическое развитие читайте в статье Паралимпиада в Сочи 2014.
Эпилептические приступы часто резистентны к терапии. С возрастом к 10 годам происходит урежение приступов.
В лечении эпилепсии при Синдроме Ангельмана используют:
- Вальпроаты (например, конвулекс ретард в дозе 20-70 мг/кг/сут).
- При миоклонических приступах и статусе абсансов — бензодиазепины (клоназепам 0,5 – 2 мг/кг/сут или фризиум 1 мг/кг/сут).
- Комбинация Вальпроата и бензодиазепина.
- При абсансах и диффузных разрядах на ЭЭГ используют Этосуксимид в дозе 20-30 мг/кг/сут.
- Комбинация Вальпроата и Этосуксимида.
- Топирамат в дозе 2- 5 мг/кг/сут.
- Ламотриджин в комбинации с Вальпроатом.
Не используют в терапии СА в монотерапии из-за возможной аггравации приступов: фенитоин, карбамазепин, окскарбазепин, вигабатрин.
Но хороший эффект в комбинации
8. При фокальных моторных приступах — комбинация Вальпроата и Тегретола (15 — 25 мг/кг/сут).
9. При кортикальном миоклонусе эффект показал пирацетам в высоких дозах 140 мг/кг/сут внутривенно капельно.
10. Кетогенная диета.
11. Препараты ноотропного действия (осторожно).
12. Седативные препараты для улучшения сна, с подбором доз.
Приведем клинический пример, послуживший поводом этой публикации.
Родители дали согласие на публикацию этого примера.
Также на сайте читайте статью о наследственном Синдроме Рубинштейна — Тейби.
В нашем медицинском центре наблюдалось несколько пациентов с синдромом Ангельмана. Обращались с первичными жалобами на эпилептические приступы. После проведенных обследований выставлялся диагноз Синдром Ангельмана.
На приём в очередной раз обратились за выпиской для МСЭ родители с ребенком 6 лет 7 месяцев.
Жалобы при обращении: Последний приступ был на фоне течения ОРВИ с подъёмом температуры до 38,5 градусов С, более года назад в 22 часа, во время бодрствования, сидел на руках у мамы: вытянулся, заведение глаз в сторону, клонико — тонические судороги, длительность 25 минут, купировался после внутримышечного введения по Скорой помощи противосудорожного препарата. Был госпитализирован в инфекционный стационар.
Ранее генерализованный тонико — клонический приступ был 2 года 10 месяцев назад (02.2011 года).
Сохраняются миоклонические судороги в момент засыпания, почти каждый вечер, от 0 до 10 раз подряд, стали менее интенсивные. Иногда мама чувствует миоклонии только своей рукой, положив руку на тело ребенка.
Сохраняется задержка психоречевого и моторного развития, отмечается легкая положительная динамика.
Самостоятельно ходит с 4 лет. Шаткая походка. Двигательная активность улучшилась. Танцует под музыку. Пытается сам есть ложкой.
Стал больше понимать, выполняет простые команды, стал хаотично возить ручкой по листу. Книжки листает, непродолжительно занимается с игрушками, смотрит мультфильмы.
Говорит 3 — 5 слов. Стал лучше понимать обращенную речь. Занимается с логопедом -дефектологом.
Периодически раздражителен. Беспокойный сон (двигательное беспокойство во время сна, поверхностный).
В терапии получает депакин — хроносферу по 250 мг на ночь (концентрация депакина в крови — 53 мкг/мл), суксилеп по 250 мг * 2 раза в день (концентрация суксилепа – 77 мкг/мл).
Анамнез. Беременность на фоне хронической гипоксии плода. Роды 2, в 37 недель, экстренное кесарево сечение. Масса при рождении 3400, длина 54 см, по Апгар 7/8 баллов.
Формула развития: голову удерживает с 2 месяцев, сидит самостоятельно с 12 месяцев, ходит у опоры с 2,5 лет. В речи к 6,5 годам – говорит 3-5 слов с 3,6 лет.
С рождения часто срыгивал, расценивали как течение дисбактериоза.
НСГ – признаки гидроцефалии.
В возрасте от 1 до 4 месяцев отмечались пароксизмы вскидывания рук перед собой, и как бы покачивание в руках. После курса Кортексина и Церебролизина, Цераксона все купировалось.
Диагноз ДЦП выставлен в 1 год 6 месяцев. Проводили курсы реабилитации, включая медикаментозное лечение прозерином, кортексином; физиолечение (электрофорез).
В 02.2012 году впервые возник приступ миоклоний с переходом во вторично — генерализованный приступ (возможно, это был миоклонический статус). На ЭЭГ — регистрировалась продолженная эпиактивность билатерально, синхронно. Задержка формирования корковых ритмов.

В 02.2012 году отмечался однократный эпизод, когда на фоне полного здоровья, после активного курса реабилитации, возникло состояние: активные миоклонии в плечевом поясе, кластерами; через 1 час появились падения назад с обмяканием, которые возникали после миоклоний. Между пароксизмами приходил в себя, то есть утрата контакта была при обмяканиях на несколько секунд. Пароксизмы продолжались по несколько секунд и длились до 1,5 суток, пока не ввели конвулекс. На фоне терапии конвулекса (35 мг/кг/сут) и дексазона пароксизмы стали уменьшаться. Приступы купированы через 3 суток.
В стационаре назначен дексаметазон №9, конвулекс по 75 мг * 2 раза в день длительно. Приступы были купированы, но отмечались побочные действия в виде интенсивного тремора.
ЭЭГ (с положительной динамикой) — регистрировалась эпиактивность по задним отведениям с короткими эпизодами билатеральной синхронизации.
С 05.2012 года конвулекс самостоятельно отменен. Приступы не отмечались.
Возобновились вздрагивания при засыпании с 07.2012 года.
На МРТ головного мозга (02.2010 года) — арахноидальная киста средней черепной ямки справа, внутренняя гидроцефалия.
МРТ головного мозга (02.2012 года) — расширение субарахноидальных щелей, киста правой височной доли (или недоразвитие).
Генетик (Москва): анализ на наследственные болезни обмена — без патологии.
Анализ на синдром Ангельмана сдан. Диагноз подтвержден в 06.2013 году.

Заключение генетика: ish del (15)(q 11.2 q 11.2)(D15S11-, SNRPN-, D15S10-, GABR3-)
Отсутствуют 4 локуса хромосомы 15 в критической области синдромов Прадера – Вилли и Ангельмана (D15S11, SNRPN, D15S10, GABR3).
Переехал на постоянное место жительства из другого региона в 2012 году.
В ПЭЦ (противоэпилептическом центре) в 12.2012 году на ЭЭГ (фон + сон) — диффузные изменения биоэлектрической активности головного мозга. Мультирегиональная эпиактивность в височно – теменно — затылочных отделах больше справа и диффузная эпиактивность. Физиологические элементы сна — слабо выражены.
В лечении: подбор депакина — хроносферы.
Состояние с положительной динамикой. Миоклонии стали реже, улучшилась походка, стал активнее, говорит несколько слогов.
В ПЭЦ 06.2013 года на ЭЭГ (фон + сон) — региональная эпиактивность в левых лобно-теменных отведениях.
С 06.2013 года был введен суксилеп. Динамика положительная.
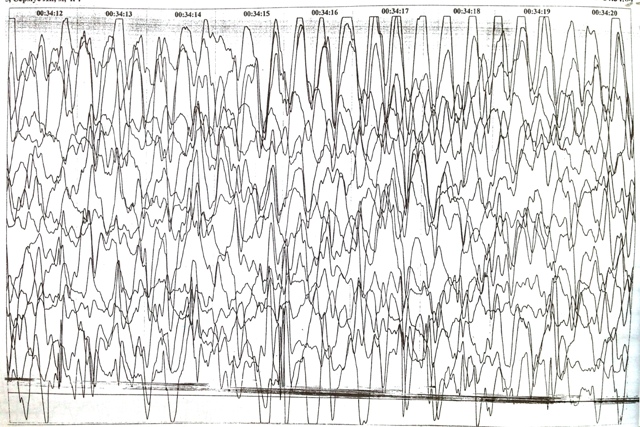
ЭЭГ (фон + сон) от 10.2013 года — региональная эпиактивность по затылочным отведениям; во сне регистрируется дельта активность по лобным отведениям с диффузным распространением.
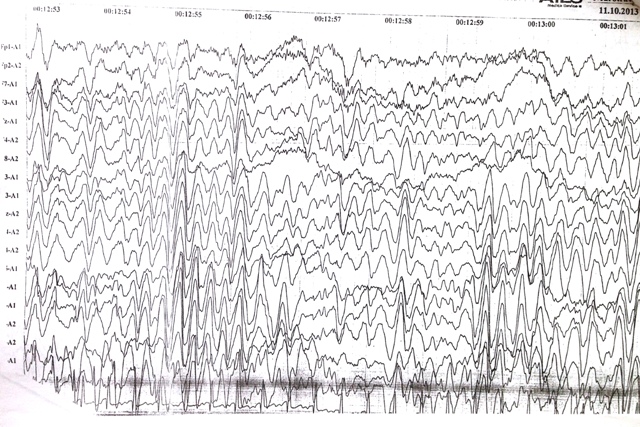
ЭЭГ (фон + сон) от 03.2014 года — диффузные изменения биоэлектрической активности головного мозга. В дремотном состоянии эпиактивности нет.
Травм, операций не было. Перенесенные заболевания — ОРВИ, ОКИ, ветряная оспа. Аллергия на сиропы, травы.
На осмотре невролога
Кожа и видимые слизистые чистые, бледные, волосы белесые.
Зев спокоен. Лимфоузлы не увеличены, отеков нет. В легких дыхание везикулярное, хрипов нет. Тоны сердца ясные ритмичные. ЧДД 20 в мин. ЧСС 84 уд в мин. Живот мягкий, безболезненный. Печень, селезенка не увеличены. Мочеиспускание свободное. Стул оформленный.
Обращает на себя внимание фенотип: маленький череп, гипоплазия средней части лица, макростомия (широкий рот), широкие межзубные промежутки, атаксия, задержка психо –речевого развития, мышечная гипотония, приступы смеха, сухожильная гиперрефлексия.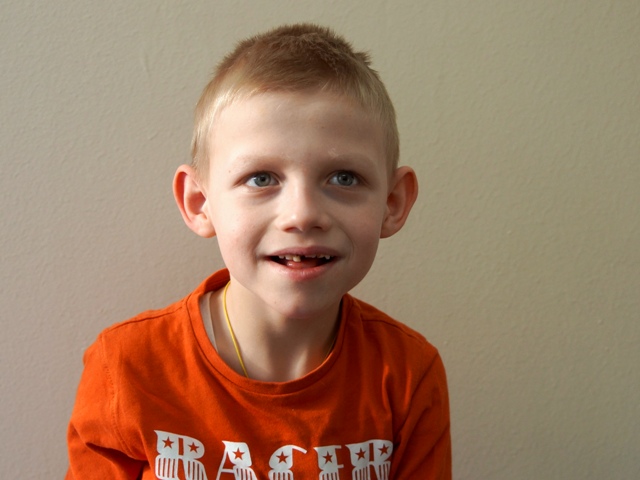
Неврологический статус
Состояние стабильное. Сознание ясное, реакция на осмотр не адекватная. Улыбчив, добродушен, легко идёт на контакт. Команды выполняет избирательно, не все понимает. Гиперактивен.
Череп микроцефальной формы (микробрахиоцефалия), окружность головы 45 см.
Глазные щели симметричны, объем движения глазных яблок полный, реакция зрачков на свет живая D=S. Лицо симметрично, носогубные складки не сглажены. Слух не нарушен. Глотает самостоятельно, не поперхивается. Язык по средней линии, высунут изо рта.
Сила мышц в конечностях 5 баллов, D=S. Тонус мышц снижен. Сухожильные рефлексы с рук и ног высокие, D=S. Патологические знаки + -.
Расстройства чувствительности нет.
Координационные пробы не понимает. Походка атаксическая, ходит на широко расставленных ногах.
Менингеальных знаков нет.
Диагноз: Миоклонический статус при непрогрессирующей энцефалопатии на фоне хромосомной патологии, синдрома Ангельмана. Органическое поражение ЦНС, атонически — астатический синдром, грубая задержка психоречевого развития, GMFCS IV уровень.
Итак, в приведенном примере мы увидели все характерные черты синдрома Ангельмана; проследили, как начинались и менялись эпилептические приступы и картина ЭЭГ; эффективность терапии. Встретив пациента с похожими симптомами, мы заподозрим и направим к генетикам на уточнения диагноза, выберем более верную тактику лечения.
Из статьи мы узнали, что представляет Синдром Ангельмана. Это патология 15 хромосомы, проявляющаяся выраженной ЗПРР и ЗМР, нарушением координации в виде марионеточной походки, аутоподобным поведением, эмоциональными особенностями (радостное лицо ангела, вспышки смеха, улыбчивость, доверчивость), стереотипиями (взмахи или похлопывания руками), эпилептическими приступами.
На видео наш пациент ребенок с Синдромом Ангельмана
